Samoprzyśpieszenie i stabilność
Samoprzyśpieszenie składników materii istnieje w różnych skalach. W mikroskali istnieje, na przykład, w każdej sieci krystalicznej. Tam każdy atom w każdym momencie poddawany jest przyśpieszeniom. Przyśpieszenia działają z różnych stron i kierują go w stronę pewnego średniego położenia równowagi pośród sąsiednich atomów. W megaskali bez samoprzyśpieszenia nie mogłyby istnieć układy planetarne.
Termin "samoprzyśpieszenie składników materii" jest bezpośrenio związany z terminem "samoorganizacja składników materii" - terminy te są powiązane ze sobą w kilku znaczeniach. Po pierwsze, proces samoorganizacji struktury jest bezpośrednim przejawianiem się procesu samoprzyśpieszenia składników struktury. Po drugie, proces samoorganizacji ma sens, jeśli istnieją co najmniej dwa składniki. Podobnie dzieje się także w przypadku procesu samoprzyśpieszenia. Inaczej mówiąc, jeśli w całym wszechświecie istniałby tylko jeden składnik, wówczas i samoorganizacja struktury (z tego jednego składnika), i samoprzyśpieszenie (tego składnika) nie istniałyby - byłyby to wówczas pojęcia bezsensowne.
Samoprzyśpieszenie konkretnego składnika jest procesem, który zachodzi w obecności co najmniej jednego dodatkowego składnika. Na tej podstawie fizycy nie mówią o samoprzyśpieszeniu - mówią oni o przyśpieszeniu, które jest powodowane przez drugi składnik. Ja nazywam to samoprzyśpieszeniem dlatego, że o naturze i o mechanizmie oddziaływań, które wymuszają przyśpieszenie składników, w rzeczywistości, nie można niczego konkretnego powiedzieć. Oczywiście, mówimy o grawitacyjnym, elektrostatycznym bądź innym oddziaływaniu, ale to są tylko nazwy, za którymi nie stoją żadne szczegółowe wyjaśnienia fizycznego mechanizmu.
Pewien dziennikarz, ukrywający się (jako autor "książki o dziwnych rzeczach", wydanej w Rosji pt. "Kniga o strannom") pod pseudonimem Byrd Kiwi (kiwi@computerra.ru , kiwibyrd@mail.ru ), nazywany najbardziej tajemniczym autorem RuNet-u, w wywiadzie (http://www.rususa.com/news/news.asp-nid-1107-catid-6) przedstawił to następująco: "...wygląda to dość smętnie, gdy jedynie najwięksi uczeni (podobni do Feynmana, który, brzydko mówiąc, pluje na pietyzm) mogą całkiem otwarcie przyznać, że w rzeczy samej współczesna nauka nie wie niczego o rzeczywistej naturze sił we wszechświecie, takich jak grawitacja czy elektromagnetyzm."
Tym niemniej, nie przeszkadza to odróżniać od siebie różnych rodzajów oddziaływań. Dlatego że odróżnianie różnych oddziaływań odbywa się na podstawie różnorodnych cech, które towarzyszą różnorodnym procesom. Po prostu, na podstawie cech mówi się o takim bądź innym oddziaływaniu.
Oczywiście, można mówić, że przyśpieszenie jest skutkiem jakiegoś oddziaływania. Ale należy pamiętać o tym, że jest to nielogiczne i nieścisłe. Ścisłość takiego stwierdzenia jest tego samego rzędu, jak ścisłość poglądu, że oddziaływanie powoduje samoorganizację struktury. Kiedy dla wyjaśnienia czegokolwiek wykorzystuje się samą nazwę, to wówczas absolutnie niczego się nie wyjaśnia.
Samoprzyśpieszenie składników i samoorganizację struktur można opisywać, biorąc za podstawę logiczne zależności i matematyczne wzory, które bezpośrednio przedstawiają przyśpieszenie składników. Oczywiście, to także nie wyjaśnia przyczyn samoprzyśpieszenia i(lub) samoorganizacji, ale pozwala modelować i jedno i drugie, i nie tylko to.
Wykorzystując matematyczne wzory na przyśpieszenie, można uzasadniać istnienie samoorganizacji i samostabilizacji struktur materialnych - to jest najważniejsze. Za pomocą takiego podejścia można modelować między innymi potencjał kontaktowy i prąd elektryczny.
Co ciekawe, modelować można na dwa sposoby.*) Do modelowania można wykorzystać składniki (cząstki), których własności są przedstawiane za pomocą przeciwnych znaków "plus" i "minus" - sposób-1, i można wykorzystać składniki, do opisania których przeciwne znaki nie są wykorzystywane - sposób-2. W pierwszym przypadku, przy dostatecznie dużych odległościach między składnikami i przy początkowych prędkościach składników równych zero, składniki o przeciwnych znakach mają przyśpieszenia, które zbliżają je do siebie, a składniki o jednakowych znakach pod wpływem przyśpieszenia oddalają się od siebie. Dlatego utarło się powiedzenie, że w sposobie-1 zachodzi wzajemne przyciąganie bądź wzajemne odpychanie składników. W drugim przypadku, przy dostatecznie dużych odległościach między składnikami i przy początkowych prędkościach składników równych zero, składniki mają tylko takie przyśpieszenia, które zbliżają ich do siebie. Dlatego utarło się powiedzenie, że w sposobie-2 zachodzi jedynie wzajemne przyciąganie się elementów.
Podkreślam, że wszystko to ma miejsce przy dostatecznie dużych odległościach między składnikami. Dostatecznie duże odległości są to takie odległości między składnikami, przy których istnieją ograniczone możliwości sformowania się stabilnych struktur materialnych. Przy takich odległościach możliwe jest istnienie stabilnej struktury. Ale do tego konieczne są dodatkowe "początkowe" prędkości składników struktury, które razem z przyśpieszeniami, i z wynikającymi z nich prędkościami, stworzą obrotowy ruch struktury. Właśnie w takiej sytuacji istnieje i działa każdy układ planetarny. W planetarnym układzie są przyśpieszenia składników, które są wynikiem obecności współtowarzyszy (planet i Słońca), i są ich orbitalne prędkości.
W przeogromnej większości układy strukturalne kształtują się przy małych odległościach między ich składnikami. W takiej sytuacji dla istnienia struktury ruch obrotowy nie jest potrzebny, chociaż, oczywiście, ruch obrotowy mikrostruktur jest tam często spotykany. Na przykład, w powietrzu molekuły gazów zderzają się ze sobą i przelatują odległość między kolejnymi zderzeniami, znajdując się jednocześnie w stanie ruchu obrotowego. W takim przypadku ruch obrotowy jest zjawiskiem ubocznym, pojawiającym się wskutek niecentralnego zderzenia molekuł.
Stabilność składników strukturalnych w atomach, molekułach i kryształach jest zorganizowania w inny sposób, niż sposób-1 i sposób-2 - ten sposób można oznaczyć jako sposób-3. Istotą tego sposobu jest to, że w nim są jakby sprzężone ze sobą zarówno przyciąganie, jak i odpychanie. Na przykład, w przypadku dwóch składników (atomów, cząstek, pól) istnieje co najmniej jedna odległość między nimi, przy której składniki mają zerowe przyśpieszenia. Przy takiej odległości mogą one pozostawać względem siebie nieruchome.
Zmiana tej odległości, oczywiście, spowodowana jakąś zewnętrzną przyczyną, skutkuje powstaniem przyśpieszenia obu składników w takich kierunkach, aby odległość ta odnowiła się. Następnie będzie istniał drgający ruch składników wokół pewnych położeń równowagi i, jednocześnie, wokół danej odległości.
Ale zmiana odległości między dwoma składnikami, która zachodzi pod wpływem zewnętrznej przyczyny, może także być zbyt wielka. Wówczas składniki albo oddalają się od siebie (szczególnie wówczas, gdy przy dużych odległościach od siebie zachowują się one jak jednoimienne składniki), albo zaczynają drgać wokół zupełnie innej (wartości) "średniej" odległości między nimi.
Można to zobaczyć, wykorzystując do tego celu komputerowy modelujący program Self-Acceleration i pliki z rozszerzeniem .ato, które są w pewnym sensie zgrupowane w "TwoTaons" bądź "TwoGravons". Jeśli załączyć, na przykład, plik TwoTaons_Cool.ato, można zobaczyć, że na początku procesu dwa składniki znajdują się od siebie w odległości 1.89664183761789 "jednostek odległości" i ich początkowe prędkości są równe zero. Po włączeniu procesu można zobaczyć, że składniki pozostają prawie nieruchome. To znaczy, one drgają, ale wielkość zmiany ich położenia w układzie współrzędnych nie jest większa od jednej tysięcznej części "jednostki odległości". Na ekranie programu widoczna jest czerwona i zielona kropka (jako odpowiedniki centralnych punktów centralnie symetrycznych pól), ale ich drgania nie są widoczne.
W pliku TwoTaons.ato początkowa odległość między dwoma składnikami zmieniła się - jest ona równa 1.8 "jednostek odległości". Także w tym pliku początkowe prędkości składników są równe zero. Ale teraz po włączeniu procesu wielkość zmiany ich położenia w układzie współrzędnych jest równa prawie jednej dziesiątej części "jednostki odległości" (czyli, teraz zmiana położenia składnika jest sto razy większa od poprzedniej) i na ekranie programu są widoczne drgania składników.
(Aby obserwować, jak zmieniają się współrzędne i prędkości składników, należy na pulpicie programu włączyć przycisk Show Listing. W Listing (tablicy parametrów, która znajduje się na pulpicie) są widoczne albo współrzędne składników, albo ich prędkości, a przełączenie na jedno bądź drugie odbywa się za pomocą podwójnego kliknięcia lewym klawiszem myszy, gdy kursor znajduje na białym polu Listing.)
Na rysunku V_E-CSField.gif są przedstawione dwie wersje rozkładu potencjału i natężenia w centralnie symetrycznym polu wzdłuż dowolnego promienia, który wychodzi z centralnego punktu pola. Natężenie pola jest równoważne przyśpieszeniu, jakie inny składnik (pole) uzyskuje w danym c.s. polu.
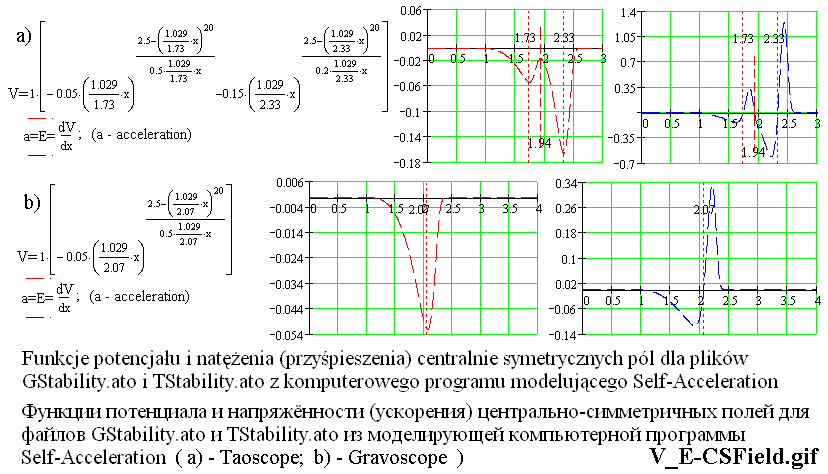
Z takich centralnie symetrycznych pól składają się dwójki składników w plikach z rozszerzeniem .ato, które są zgrupowane w "TwoTaons" lub w "TwoGravons", i z nich składają się wieloskładnikowe struktury, które są zapisane w plikach "Gstability" i "TStability". Różnica jest taka, że na rysunku V_E-CSField.gif przyjęto liczbową wartość parametru A=1 (to liczba "1" przed kwadratowymi nawiasami), a parametr A centralnie symetrycznych pól, które znajdują się w plikach "Gstability" i "TStability", mają liczbowe wartości równe: 200, 800, 20000, 80000. (W komputerowym programie Self-Acceleration w wymienionych plikach parametr A dla różnych c.s. pól jest oznaczony jako A2 albo A3, a w innych plikach, o których będzie mowa poniżej, bywa on także zapisany jako A1.)
Potencjał kontaktowy i elektryczność
W plikach "Gstability" i "TStability" stabilne struktury składają się z szesnastu składników. Osiem składników ma parametr A2=200 (w innej wersji 20000), a pozostałe osiem składników ma parametr A3=800 (w innej wersji 80000). Przedstawione w plikach struktury przypominają strukturę, która istnieje na styku dwóch pierwiastków chemicznych i, podobnie jak ta struktura, tworzą potencjał kontaktowy.
Jest to widoczne wówczas, gdy do takiej modelowej struktury dopisać dodatkowo jeszcze trzeci tym c.s. pól, które mają, na przykład, parametr A3=0.1. Te dodane pola zachowują się jak elektrony na styku dwóch pierwiastków chemicznych. Wówczas przejawia się zarówno potencjał kontaktowy, który istnieje na styku dwóch odmiennych polowych struktur (z parametrami c.s. pól A2=200 i A3=800, albo odwrotnie, A2=800 i A3=200), jak i powstaje ukierunkowany ruch pewnej liczby dodanych c.s. pól, który w tym programie jest odpowiednikiem prądu elektrycznego.
Modelowanie potencjału kontaktowego i prądu elektrycznego jest równoważne przedstawieniu mechanizmu pracy termoelementu. Mechanizm ten można obejrzeć, wykorzystując grupy plików z rozszerzeniem .ato: "GCoCoPo", "GHoCoPo", "TCoCoPo", "THoCoPo". Pierwsze litery w nazwach plików G bądź T oznaczają, że podczas pracy z danym plikiem w programie Self-Acceleration należy włączyć albo przycisk Gravoscope, albo przycisk Taoscope. Następne litery są skrótem od słów "cold contact potential" (zimny potencjał kontaktowy) i "hot contact potential" (gorący potencjał kontaktowy).
Za pomocą modelującego programu Self-Acceleration można przedstawić dwie wersje stanu potencjału kontaktowego. Można przedstawić "zimną" wersję, w której oba termoelementy - ten, który jest widoczny na ekranie programu, i ten, który na ekranie nie jest przedstawiony - mają jednakowe temperatury. I można przedstawić "gorącą" wersję, w której oba termoelementy mają różne temperatury. Oczywiście, należy przy tym wyobrażać sobie, że struktura, która jest przedstawiona na ekranie programu, składająca się z trzech rodzajów c.s. pól, jest maleńkim fragmentem zamkniętego obwodu elektrycznego; pozostałą cześć tego obwodu należy utrzymywać w wyobraźni.
W przypadku "zimnego potencjału kontaktowego" oba kontakty mają jednakowe temperatury. Dlatego na ekranie programu można obserwować jedynie pewne skupienie składników, modelujących elektrony. Skupienie składników-elektronów zachodzi w obszarze struktury, gdzie parametr c.s. pól A=800, i tam ich ruch jest bardziej ożywiony. Oczywiście, dzieje się tak z tego powodu, że tam zarówno różnice potencjałów, jak i uzyskiwane przez składniki-elektrony ich przyśpieszenia są większe, niż w obszarze struktury, gdzie parametr c.s. pól A=200.
Tego rodzaju sytuacje można obserwować po naciśnięciu przycisku "cold" contact potential, za pomocą którego są załączane pewne ograniczenia dla ruchu składników-elektronów, ale także można obserwować po naciśnięciu na przycisk AtomStand, kiedy żadne ograniczenia na ruch składników-elektronów nie są nakładane. Kiedy na pulpicie programu w aktywnym stanie pozostaje przycisk AtomStand, wszystkie składniki struktury, które biorą udział w procesie, poruszają się zgodnie ze swoją naturą - każdy składnik ma przyśpieszenie, które zależy od pozostałych składników; żadne dodatkowe warunki dla ich ruchu nie istnieją.
Ograniczenia, o których tu mowa (przy aktywnym przycisku "cold" contact potential), polegają na tym, że składniki-elektrony poruszają się jedynie w pewnym obszarze przestrzeni, nie oddalając się zbytnio od podstawowej struktury, i uzyskują prędkości, które nie przekraczają pewnej wartości. W ten sposób za pomocą przedstawionych ograniczeń są modelowane dwa parametry, które są spotykane w rzeczywistych warunkach pracy termoelementów - opór elektryczny i temperatura. Ograniczenia te nie pozwalają, aby składniki-elektrony uzyskiwały zbyt duże prędkości i żeby wskutek tego zachodziła ich emisja do "nieograniczonej" przestrzeni.
Po naciśnięciu na przycisk "Hot" contact potential (czyli, po jego aktywacji), oprócz wyżej opisanych ograniczeń, które istnieją także obecnie, załączane jest (znajduje zastosowanie) jeszcze jedno dodatkowe rozwiązanie, które daje możliwość modelowania pracy termoelementów, kiedy ich temperatury są różne.
Jest oczywiste, że kiedy jeden z termoelementów w obwodzie ma większą temperaturę, to oznacza to, że jego składniki-elektrony poruszają się w bardziej ożywiony sposób, a jego potencjał kontaktowy powiększa się i jest większy od potencjału kontaktowego chłodniejszego termoelementu. Różnica wielkości potencjałów kontaktowych gorącego i zimnego termoelementu jest przyczyną ukierunkowanego ruchu elektronów, czyli powstania prądu elektrycznego.
W modelującym programie Self-Acceleration przepływ prądu składników-elektronów jest realizowany w następujący sposób. Kiedy składnik-elektron na ekranie dochodzi do lewej bądź prawej ścianki "ograniczonego obszaru", wówczas, oczywiście, jego prędkość jest skierowana w stronę ścianki. W tym momencie znika on w tym miejscu i pojawia się przy przeciwległej ściance, posiadając wcześniejsze parametry prędkości, czyli teraz jego prędkość jest skierowana w stronę "ograniczonego obszaru". Teraz może on przelecieć przez cały obszar i może powtórzyć się akt zniknięcia przy jednej ściance i pojawienia się przy drugiej, przeciwległej ściance.
O składniku-elektronie, który dolatuje do ścianki, znika i pojawia się przy przeciwległej ściance, można powiedzieć, że w pewnym sensie gra on podwójną rolę. Do momentu zniknięcia-pojawienia gra on "swoją" rolę - w momencie zniknięcia można o nim myśleć, że on popłynął sobie dalej do obwodu elektrycznego. A po upływie momentu zniknięcia-pojawienia gra on rolę nowego składnika-elektronu, który przybywa do "ograniczonego obszaru" termoelementu z obwodu elektrycznego.
__________________________________
*) Mowa jest o modelowaniu za pomocą dwóch sposobów, pomimo że wspomina się o sposobie-1, sposobie-2 i sposobie-3. Na wymienione tutaj (i stosowane w programie Self-Acceleration) dwa sposoby modelowania stabilnych struktur materii składają się własności sposobu-1 i własności sposobu-2, i do każdej grupy własności są dodane własności sposobu-3. Przy modelowaniu za pomocą tych dwóch sposobów własności sposobu-1 bądź sposobu-2 przejawiają się przy dużych odległościach, a przy małych odległościach w obu sposobach przejawiają się własności sposobu-3. Te dwa sposoby można by oznaczyć jako sposób-(1i3) i
sposób-(2i3).
Komputerowy program modelijący Self-Acceleration i pliki z rozszerzeniem .ato
Fragment kodu programu Self-Acceleration - Ograniczenia ruchu c.s. pól
____________________________________
Odkrycie fizyki na trzecie tysiąclecie - http://pinopa.republika.pl/
Źródło: http://www.pinopa.republika.pl/SelfAcceleration_pl.html